Would This Have Flagged COVID?
Thanks to Mike McLaren, Dan Rice, James Kremer, and Alessandro Zulli for conversations that led to this post, and to Chris Doering, Mike McLaren, and Evan Fields for review.
When we explain the algorithms we use to analyze untargeted metagenomic sequencing data people always ask: would this have flagged COVID? I really like this question because we should at least ensure we would have detected the last pandemic, but especially because digging into it gives an opportunity to cover an important determinant of detection sensitivity and explain how it might apply to future pathogens.
Consider a system like CASPER. To generate an alert, the system needs to perform two key functions:
- Physical: Sequence a read from the pathogen.
- Algorithmic: Recognize that the read indicates a problem.
Insensitivity in the physical stages is a significant concern: you might fail to generate the necessary sequencing reads if you don’t happen to have any sick people shedding into your samples, your sequencing methods aren’t sensitive enough to this pathogen, your libraries don’t have enough complexity, or you’re not sequencing deeply enough. While we’ve put substantial work into these questions, here we’ll focus on understanding the sensitivity of the algorithmic portion.
When researchers in China originally identified the SARS-CoV-2 genome (Wu et al. 2020), they had untargeted metagenomic sequencing data. Because the data was from bronchoalveolar lavage fluid sampled from a single very sick patient, 22% of the sequencing reads were from SARS-CoV-2 (SCV2). With wastewater, however, we found that fewer than 0.00002% of reads matched SCV2 when 1% of people were infected (Grimm et al. 2025). Where Wu et al. could assemble the genome, we instead need to make initial assessments from individual sequencing reads. How would our read-based algorithms have fared against SCV2?
Clades of Concern (CoC), which compares sequencing reads to known pathogens, is our algorithm that would have been most sensitive to SCV2. When SCV2 first started spreading it wouldn’t have been in our databases yet, and the closest available genome would have been bat SARS coronavirus ZC45 (MG772933) uploaded in early 2018. You can see SCV2 aligns well with ZC45, with the exception of the first half of the spike protein gene (21,697 to 23,074), which contains the receptor binding domain:
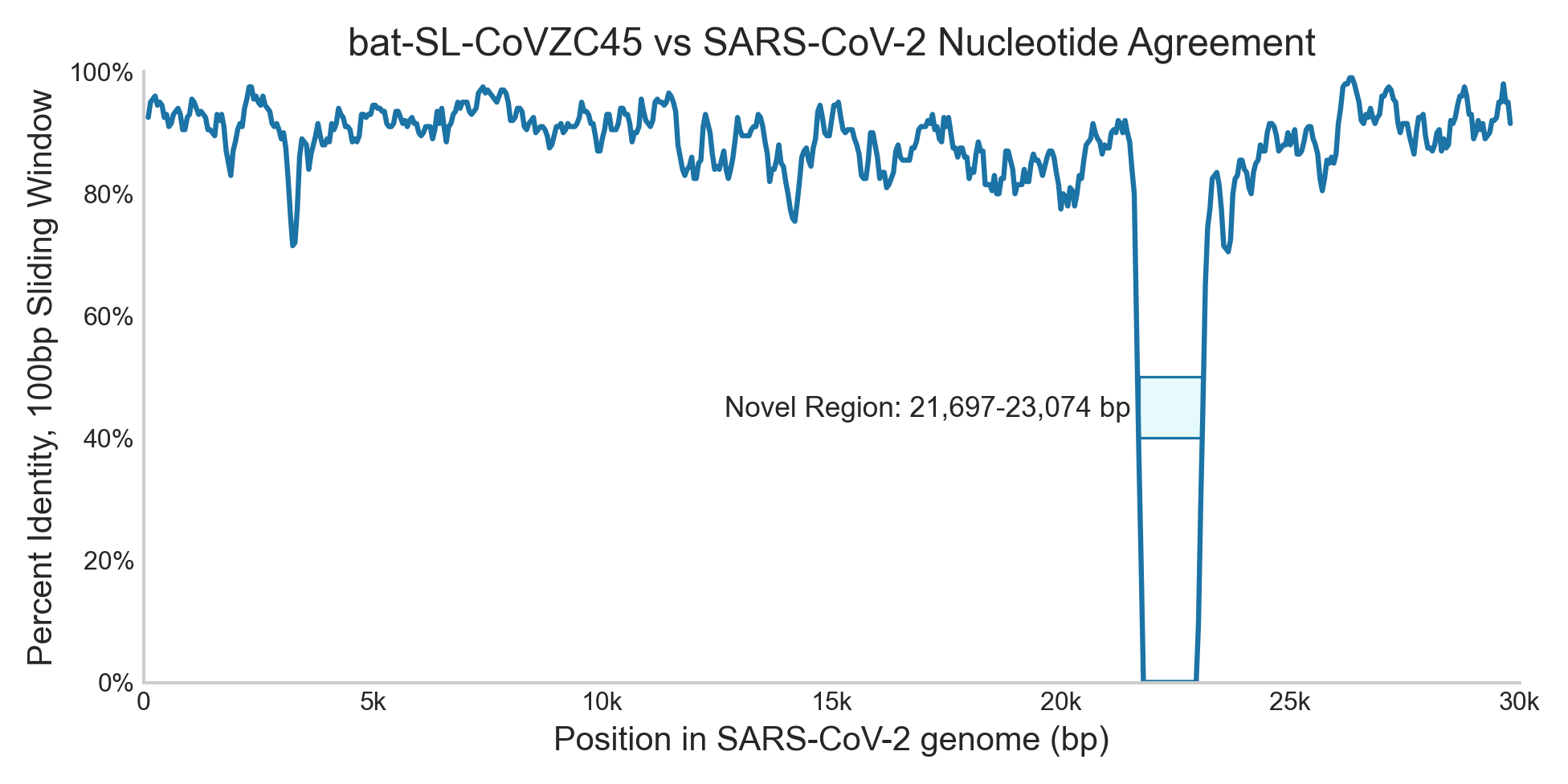
The SCV2 genome is 29,903 base pairs (bp) and this missing section is 1,378 bp, which makes the gap about 5% of the total genome. CoC would have flagged reads from the remaining 95% of the genome as suspiciously close to some kind of SARS-like pathogen, though it would have been unable to identify reads falling entirely within the novel region. Even with that blind spot, however, so much of the genome is informative that we can confidently say this would have flagged COVID.
If CoC were unavailable, however, it would likely have been flagged by Chimera Detection1 (CD). This algorithm flags sequencing reads where only a portion matches a known pathogen. Since there are discontinuities in the alignment at positions 21,697 and 23,074, CD would likely have flagged reads that crossed those points. In our sequencing the median read is 170bp after adapter trimming, and you probably need at least 30bp on either side of a discontinuity to reliably identify that something is unexpected. This means only the ~1% of reads that contain a discontinuity are informative.2
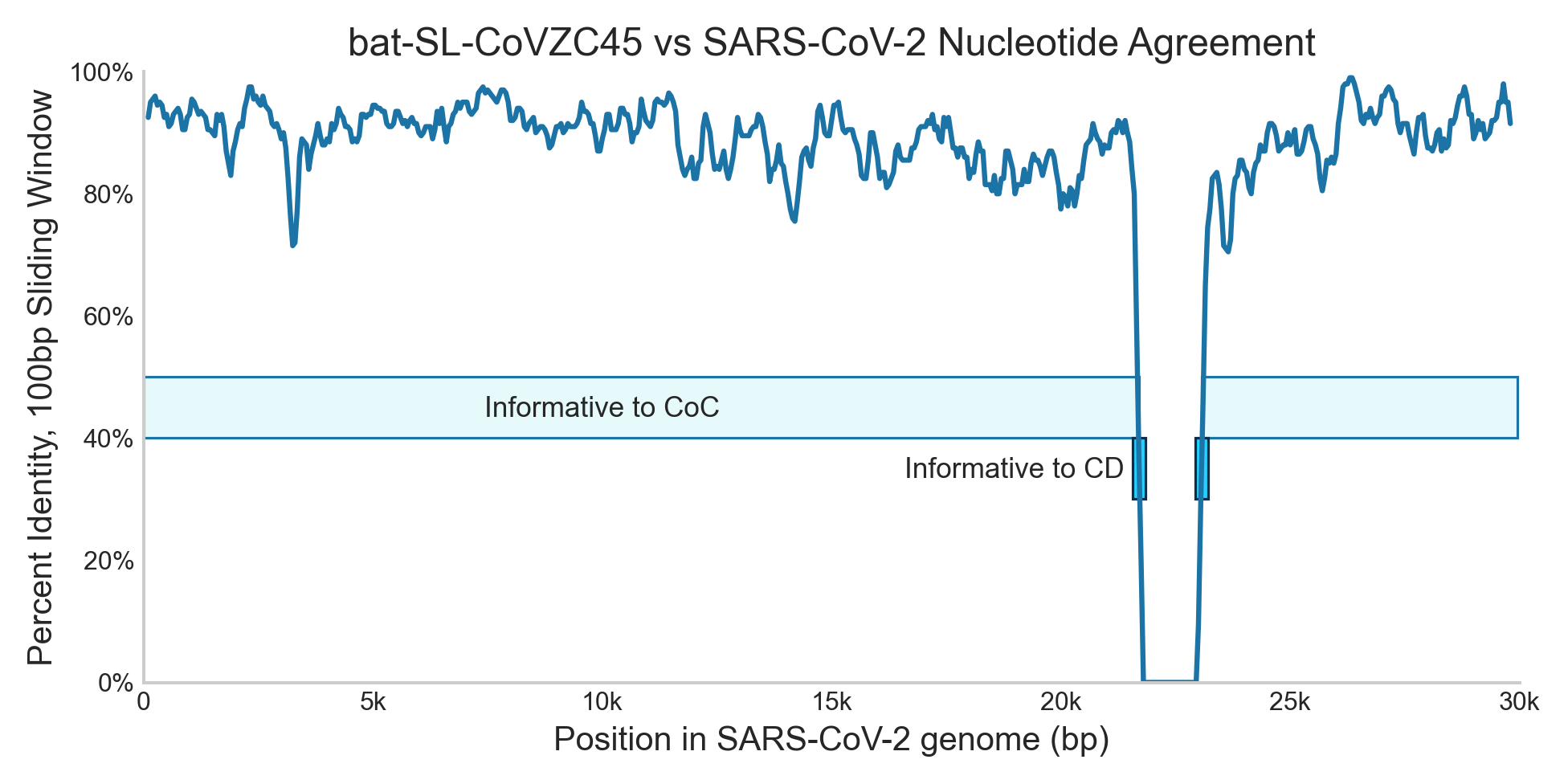
This contrast, where ~95% of reads from a pathogen genome are informative to CoC but only ~1% are informative to CD, illustrates one of the most important ways in which detection algorithms vary: what fraction of reads are useful? For example, say we’re comparing two different algorithmic approaches for identifying de novo engineered viruses:
Identify reads which code for proteins that a model predicts would bind to mammalian host cell receptors.
Identify reads with no sequence similarity to anything normally observed in this sample type, and assess them to see whether they look potentially viral.
Neither of these is an easy method to implement, let alone get working reliably as part of a cost-effective detection system processing hundreds of billions of reads weekly with acceptably few false positives. Nevertheless, they differ dramatically in their theoretical sensitivity. To method A, only a small fraction of potential reads from a given pathogen would be informative, because most of a viral genome codes for internal machinery rather than surface proteins that mediate host cell entry. To method B, however, most of the pathogen’s sequence is informative, allowing detection far earlier than with method A; this is a strong reason to prioritize B.
Returning to the motivating question, both CoC and CD would likely have flagged COVID, but CD would have needed two orders of magnitude more sequencing reads. This difference in sensitivity is a critical consideration in modeling the cost-effectiveness of real-world deployments, and in prioritizing algorithm development.
Footnotes
We’re not making any claim about SCV2’s origin here: even though CD was designed to flag engineered pathogens, it can flag a range of changes. These changes could be from natural recombination, strongly divergent selection pressure, or engineering; it’s all the same to the algorithm.↩︎
If reads are 170bp and the genome is 29,903bp then there are 29,734 possible 170bp sequencing reads (29,903 - 170 + 1). Similarly, if you need 30bp of context on each side of the junction then there are 111 possible 170 bp sequencing reads (170 - 30 - 30 + 1) that contain the junction with enough context to recognize it. With two junctions that’s 222 out of 29,734 possible reads, or 0.7%. Note that this is a rough calculation and not a simulation: actual reads have a range of lengths, and sequencing coverage isn’t uniform along a genome.↩︎