Metagenomics: A solution to PCR failures
Note: The Nucleic Acid Observatory (NAO) is now SecureBio Detection.
Introduction
Environmental surveillance of pathogens has become an integral part of public health response and epidemiology. Countries all over the world, including the US, Switzerland, most EU countries, South Africa, and Brazil are using wastewater-based epidemiology to inform their public health responses to endemic seasonal pathogens as well as emerging infectious diseases. Many of us are familiar with how wastewater-based epidemiology has contributed to SARS-CoV-2 responses, but it’s now being used to respond to re-emerging pathogens, including H5N1, measles and dengue. All of these efforts represent significant contributions to public health and share another common feature: they use targeted molecular methods, mainly polymerase chain reaction (PCR) based assays. These methods remain the gold standard for rapid and cost-effective monitoring of known targets. As an inherent limitation of the technology, however, they’re prone to pitfalls that metagenomic approaches circumvent.
For the unfamiliar, targeted molecular methods such as PCR are the biological equivalent of Ctrl-F. They look for a specific “word” (nucleic acid sequence), and copy it until there’s enough for us to see. There are clear advantages and disadvantages to this approach: on the one hand, it’s fast and relatively cheap; on the other hand, you need to know exactly what you’re looking for, and you’ll never know what else is hiding in your sample.
If PCR is Ctrl-F, metagenomics is reading the document. Instead of checking for a specific nucleic acid sequence, you read a broad subsample of everything. As much DNA and RNA as possible is read, giving you the exact sequences of As, Cs, Ts, and Gs that are present. Just as with reading a document, this can be slower and more laborious, but it helps us understand the bigger picture and pick up on things we otherwise would have missed. A metagenomics approach lets us see the usual suspects, as well as proactively detecting novel, unknown, and emerging threats.
As part of WastewaterSCAN and the Yale SARS-CoV-2 wastewater surveillance program, I’ve designed and tested dozens of PCR assays for use in wastewater-based epidemiology, including ones for wild-type measles, H5N1 markers, and antibiotic resistance genes. These methods work very well for reactive public health purposes, but along the way, we’ve encountered difficulties that metagenomic approaches could easily circumvent.
Previous challenges with PCR based detection
SARS-CoV-2: HV69-70 and S gene dropout
Back in 2020, when SARS-CoV-2 was still new, many competing assays were available. The CDC published a set of assays (N1/N2/N3), and these were used initially before the N3 assay was dropped for cross-reacting with the original SARS. The N1 and N2 assays remained widely used in academic and clinical settings. Eventually, though, one specific platform became incredibly widespread for clinical diagnoses: Thermo Fisher’s TaqPath three-gene multiplex targeting the ORF1ab, N and S genes. The usage of three different targets proved to be a good precaution, as demonstrated in the winter of 2020.
In November of 2020, the national lighthouse laboratory network in the UK noticed an increase in SARS-CoV-2 tests where the ORF1ab and N gene were positive, but the S gene was negative. UK public health began investigating, and in late December, published a technical briefing indicating that this dropout wasn’t a technical error, but evidence of a novel variant of SARS-CoV-2 rapidly spreading. SARS-CoV-2 Alpha (B.1.1.7) was the first SARS-CoV-2 variant to show much higher transmissibility than the original, and tracking it became a public health concern around the world (Figure 1).
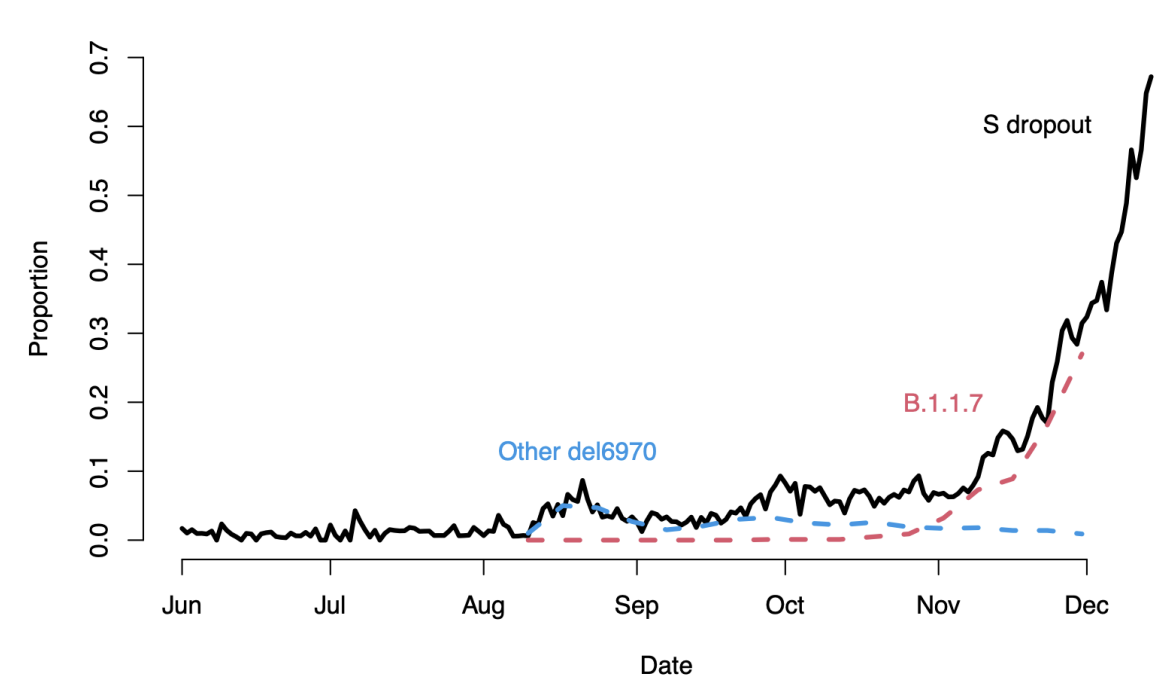
Source: UKPHE, Technical Briefing. Figure 1: The proportion of positive SARS-CoV-2 tests that showed S gene dropout in the UK during winter of 2020. The black line is S gene dropout, while the red line shows the proportion of human SARS-CoV-2 sequences identified as the B.1.1.7 variant.
The Alpha variant quickly spread worldwide, with the S gene dropout following it. The S gene dropout was caused by a mutation of the SARS-CoV-2 genome called HV69-70, allowing us to track the spread of Alpha variant by monitoring when the assay failed. This first variant highlighted a gap in our existing surveillance systems. By the time the technical briefing was released on December 21, 2020, Alpha already accounted for more than 50% of circulating SARS-CoV-2. Despite knowing these mutations might happen, we weren’t ready to test for a rapidly mutating virus.
At the time, I was running the Yale SARS-CoV-2 wastewater surveillance program. With the benefit of foresight from the UK’s lighthouse labs, I developed an assay that specifically targeted the HV69-70 deletion, enabling us to directly quantify the proportion of Alpha in our wastewater samples. We recognized that this was a stopgap solution; after all, nothing was stopping a different mutation from popping up that would render our assay useless. We worked with a team to apply a sequencing-based approach based on ARTIC amplicon sequencing and demonstrated that this approach could work in wastewater, closely tracking molecular results from the assay and clinical results.
Even then, this approach only worked for SARS-CoV-2 and its variants. We would be blind if a new virus were to show up, throwing a monkey wrench into our surveillance plans .
Mpox Clade I, II and Ib
Mpox, caused by Orthopoxvirus monkeypox virus, is in the same genus as smallpox and causes rash, blisters, and fever. In the summer of 2022, Mpox Clade II, previously found only on the African continent, reached the United States. Public health mounted a significant response, tracking thousands of cases and setting up vaccination campaigns for at-risk populations. All of this tracking, including wastewater testing, was done using PCR.
These efforts were successful, but there was worry that Mpox Clade I, a more severe strain of the virus, with a significantly higher mortality rate, would spread similarly. Public health and academic researchers alike had assays ready and had learned our lessons from previous experience.
What no one expected was that Clade I would mutate significantly, cutting out an entire segment right where the assay targeted it (Figure 2).
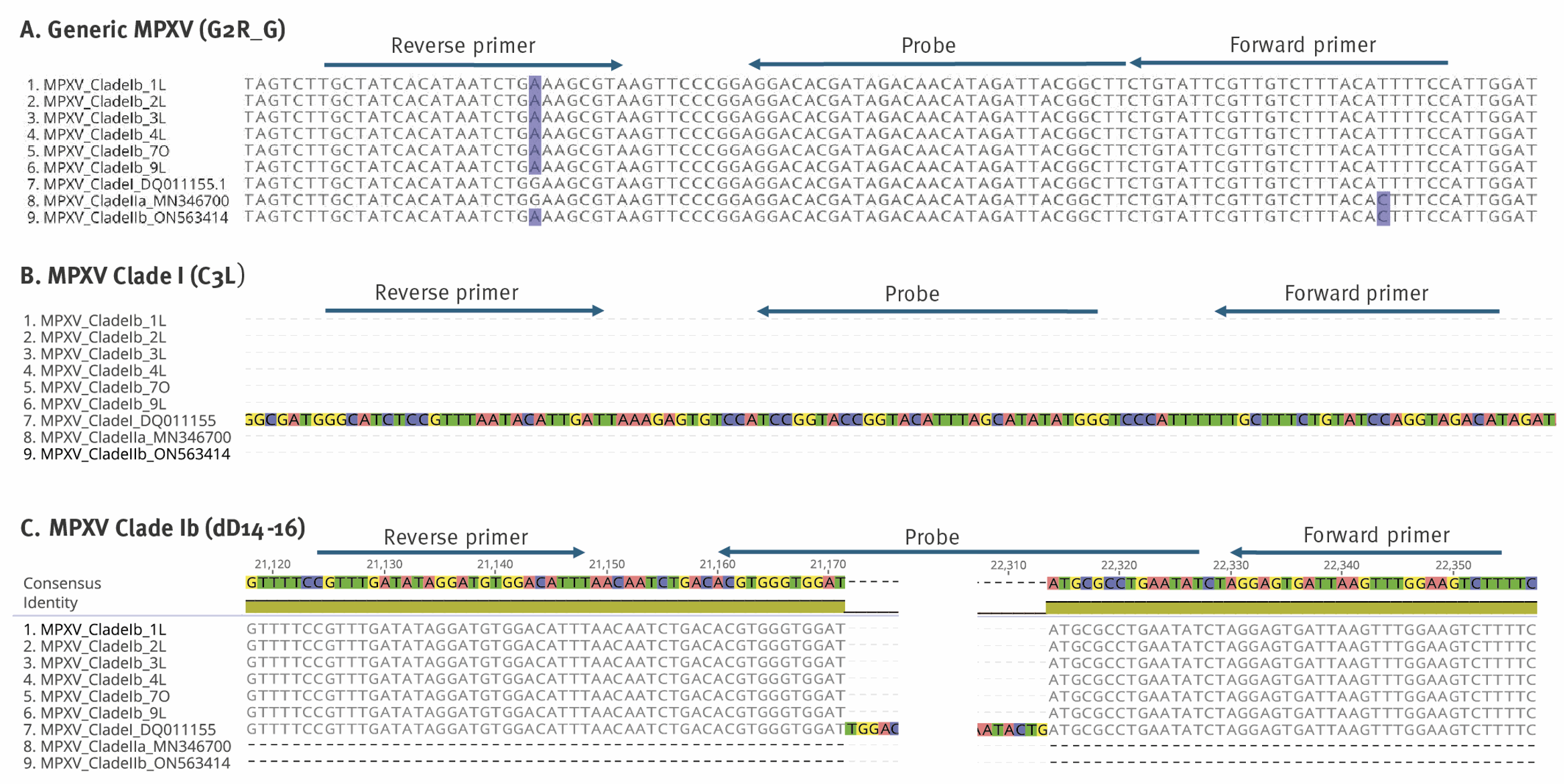
Source: Schuele et al., Eurosurveillance 2024.
Figure 2: Sequences of the mpox virus, showing how the virus had mutated. Panel B shows the previous Clade I genome, and where the primers and probes would bind. Panel C shows the new Clade Ib, highlighting the missing section where the previous probe would no longer bind.
Genetic evidence suggests that this divergent Clade Ib lineage may have been circulating undetected as early as 2011, but it was not formally identified until January 2024. The outbreak had already begun in September of 2023, meaning four months had passed before the strain was identified as Clade Ib. Targeted assays were subsequently developed and, after extensive testing, deployed for both clinical and wastewater surveillance at WastewaterSCAN. The first validated PCR assay was not published until August 2024.
While transmission of Clade Ib remained thankfully low, these delays could have been costly: a virus’ doubling time is measured in days, not months. A metagenomic approach would have circumvented these delays, as well as perhaps allowing for the earlier detection and distinction of Clade Ib from related strains.
Measles and the vaccine
Measles is a complicated virus, particularly in environmental surveillance. All measles vaccines are based on attenuated measles virus strains: a lineage of the virus that has been severely weakened but still generates an immune response.
Unfortunately for environmental surveillance efforts, both the wild type measles virus and the vaccine are shed significantly by people. This isn’t a problem for clinical purposes, but presents a significant challenge when working with wastewater samples. The existing measles PCR assays were general, focused on catching all the different strains of measles (wild-type) that exist, and as a result, would also catch the vaccine. Still this was the best that was available and we needed to begin tracking measles, so WastewaterSCAN began using these assays in wastewater as cases began to rise. It quickly became clear we needed something better. When we had detections, our meetings with public health went a little like this:
“We found measles in wastewater”
“How do you know it’s not the vaccine?”
“Well, we don’t but we’re only finding it in areas with measles cases so far.”
“That’s not actionable.”
This was frustrating, but correct on their part. Our detections had no certainty, and public health cannot afford to cry wolf.
Labs began to investigate a range of approaches. Some tried to distinguish the wild type from the vaccine based on differences in amplitude (how bright the DNA glows), but since fluorescence, particularly in wastewater, can be affected by a variety of factors, this introduced another source of uncertainty.
Being part of this response, I came across a paper by Roy et al. that developed assays to identify the vaccine and ignored the wild-type measles virus. This allowed us to identify a primer/probe set that inverted this, targeting just the wild-types B, D, and H. The assay was tested, and it worked incredibly well. We could now let public health know with certainty, and they could respond appropriately.
This PCR assay is an invaluable tool for sensitive detection of wild-type measles and for identifying outbreaks as they happen across the country. It did, however, take quite a bit of time and scientific effort to develop and test. Metagenomic sequencing would have circumvented the issue once again. Reads could have been investigated in detail, allowing us to determine whether it was the vaccine or wild-type and even telling us which strain of measles it represented.
That’s exactly what SecureBio and our CASPER collaborators did.
In a wastewater sample from late October 2025, sequenced by our collaborators in Marc Johnson’s lab, we detected several reads of Morbillivirus hominis: measles. We investigated these reads, mapping each one against recent genomes of infected persons and the genome of the vaccine. What we found was that the reads were significantly different from the vaccine strain, but perfectly matched the measles circulating in the population (Figure 3).
By leveraging sequencing data, we’d not only found measles, but been able to conclusively show it matched the wild type. No additional testing, no development of specific assays. The information was all there.
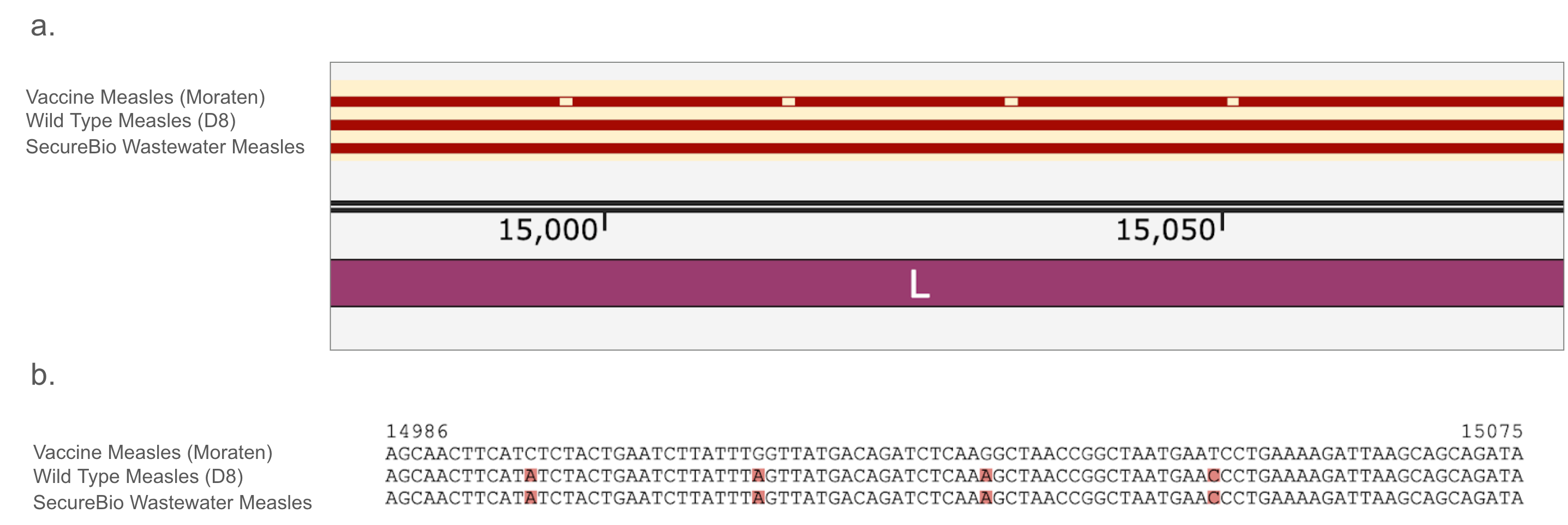
Figure 3: Genomic confirmation of wild-type measles in wastewater. Part a shows the vaccine strain compared to wild type and SecureBio’s wastewater detection along the measles genome. Mismatches between the vaccine and the latter two are shown as blank boxes. Part b shows more details of the alignment, showing that not only did the wastewater not match the vaccine, it perfectly matched the circulating wild type, with the same mutations in the same locations. Mismatches are highlighted in red.
Influenza, the ever-mutating virus
Influenza A, prior to SARS-CoV-2, was the single most deadly respiratory virus in the world. A major reason for this is its high degree of mutability. Each year, some of the world’s leading virologists, immunologists, and influenza experts get together to predict the four (three now, as we eradicated influenza B Yamagata during COVID) influenza strains that will spread this year. Sometimes they get it right, and sometimes not quite (Subclade K).
When dealing with environmental samples, I’ve encountered this influenza issue in various ways. As part of WastewaterSCAN, we ran several influenza assays on wastewater samples. One targeted a conserved region generic to all influenza on the M gene, while 3 other assays allowed us to subtype our influenza signal (H1, H3, H5). The H gene, which stands for hemagglutinin, encodes the protein that allows the influenza virus to infect human cells. As a result, this region is highly variable, constantly battling with our immune system to escape it.
In late 2024, the H3N2 influenza H gene mutated significantly. This meant that, despite rising influenza M gene levels, the subtyping assays could no longer ‘see’ the virus: there didn’t seem to be a corresponding rise in H1 or H3. This represents a fundamental limitation of targeted surveillance: when the biology changes, the tool stops working. In this case, the signal was also partially masked by a simultaneous rise in H5, due to the avian influenza outbreak. Eventually, this problem was caught, new assays were designed, and samples were rerun. This also led to the design of a process in which assays were checked monthly against the most up-to-date influenza sequences from GISAID, allowing WastewaterSCAN to catch any potential mismatches much earlier.
In the summer of 2025, WastewaterSCAN observed a spike in influenza A concentrations comparable to those during peak winter influenza season (Figure 4). There was no associated spike of clinical cases. We tested wastewater using PCR assays for markers of influenza H1, H3, H5, H7, and H9, but did not detect any.
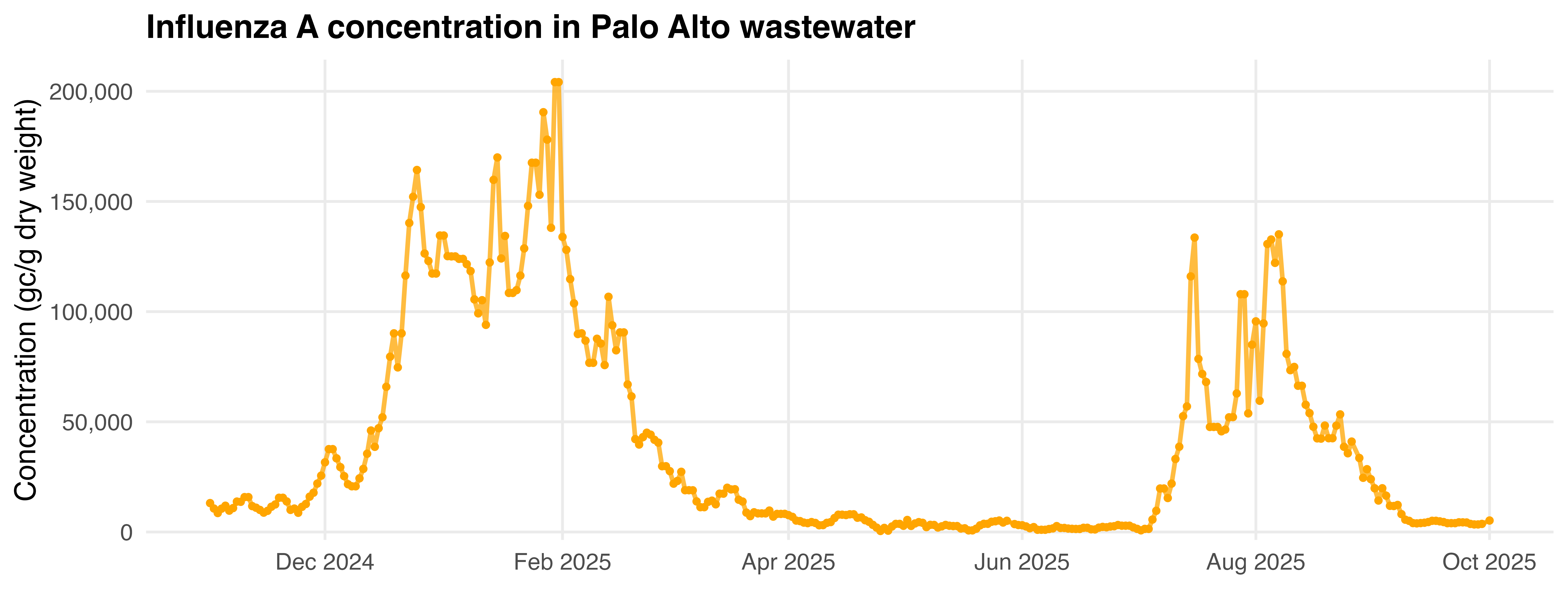
Source: WastewaterSCAN Dashboard.
Figure 4: Concentrations, in gene copies per gram of dry weight, of influenza A in Palo Alto wastewater from WastewaterSCAN. The left side of the graph shows the typical winter influenza season, between November 2024 and March 2025 and with thousands of associated clinical cases. The right side of the graph shows the atypical summer influenza spike which had no associated clinical cases.
As you may have guessed by now, myself and the team at WastewaterSCAN solved this puzzle with sequencing.* It was… well, the preprint should be out soon. The PCR panel did exactly what it was designed to do, checking for specific targets, but because this was an unusual subtype, outside the standard panel, it remained invisible. It was sequencing, and some careful bioinformatics, that allowed us to identify the exact subtype of influenza A present in the wastewater and inform local public health.
Metagenomics: the advantages and challenges
Until recently, the depth of sequencing required to detect pathogens in wastewater with an untargeted approach was prohibitively expensive, making PCR the only viable option for biosurveillance. Costs have been falling rapidly, however, and metagenomics is becoming a scalable all-in-one solution. It can be used to monitor the epidemiology and evolution of existing pathogens such as SARS-CoV-2 and influenza, give you early warning of emerging pathogens such as mpox and measles, and identify novel, potentially engineered pathogens. As the cost, speed, and precision of Next Generation Sequencing continues to improve, we need to keep building the systems and know-how to fully leverage this technology.
This is not to say PCR should be ignored. As culture-based methods (growing a pathogen) have continued to be used as a complement to PCR in research and clinical settings, so can PCR continue to complement metagenomics in developing robust public health and biosafety systems. Metagenomics can allow us to identify threats to human health before we know what we’re looking for, and once those threats are identified, rapidly developed PCR assays will be key to rapid and cost-effective response.
About the author: Alessandro Zulli is a research scientist and Zephyr project lead at SecureBio. Previously, he led the Connecticut SARS-CoV-2 monitoring program at Yale University (2020-2023), and was a senior research engineer for WastewaterSCAN at Stanford University (2023-2025).
**Special thanks to Alex Jaffe for his work on this.*